
Vol.4 エキスパート版 血管内皮細胞とグリコカリックス
アイコンが表示されている用語は記事内でも解説が確認できます。
用語解説1.正常/静的均衡状態
サマリー
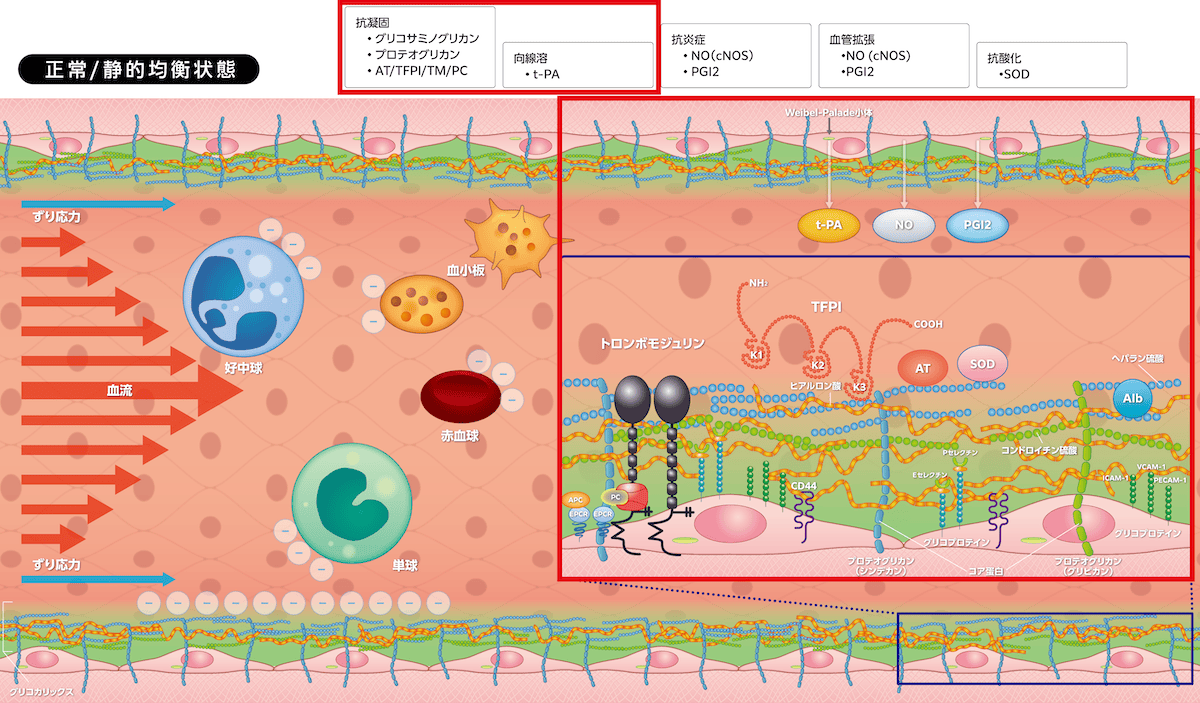
血管内皮細胞とグリコカリックスは血管の静的均衡状態(quiescence)を維持しています。グリコカリックスは毛細血管、小動静脈、中・大血管、赤血球、白血球、血小板、気道・消化管・尿細管上皮細胞などに発現し、グリコプロテインおよびプロテオグリカン、グリコサミノグリカンで構成されます。血管内皮細胞とグリコカリックスには3つの生理作用があり、①
アンチトロンビン
アンチトロンビン:antithrombin(AT)
トロンビンを中和して凝固を阻害する抗凝固因子(セリンプロテアーゼインヒビター、セルピン)。
・組織因子経路インヒビター(TFPI)
組織因子経路インヒビター:tissue factor pathway inhibitor(TFPI)
TF/FVIIa、FVa、FXaを阻害する血液凝固制御因子。
・トロンボモジュリン/
プロテインC
プロテインC:protein C(PC)
抗凝固因子の1つで、トロンビン/トロンボモジュリン複合体により活性型PC(APC)となる。
による抗凝固作用、②
t-PA
t-PA:tissue-type plasminogen activator
組織型プラスミノゲンアクチベータ。プラスミノゲンをプラスミンに変換する線溶系の開始因子。
による向線溶作用、③内皮型一酸化窒素合成酵素(eNOS)/
NO
NO:nitric oxide
一酸化窒素。
・プロスタグランジン(PG)I2
プロスタグランジンI2: prostaglandin I2(PGI2)
血管内皮細胞で合成され、血小板凝集抑制と血管拡張を起こす。
・SOD
SOD:superoxide dismutase
細胞内に発生した活性酸素を分解する酵素。
・グリコサミノグリカン陰性荷電による抗炎症作用が挙げられます。
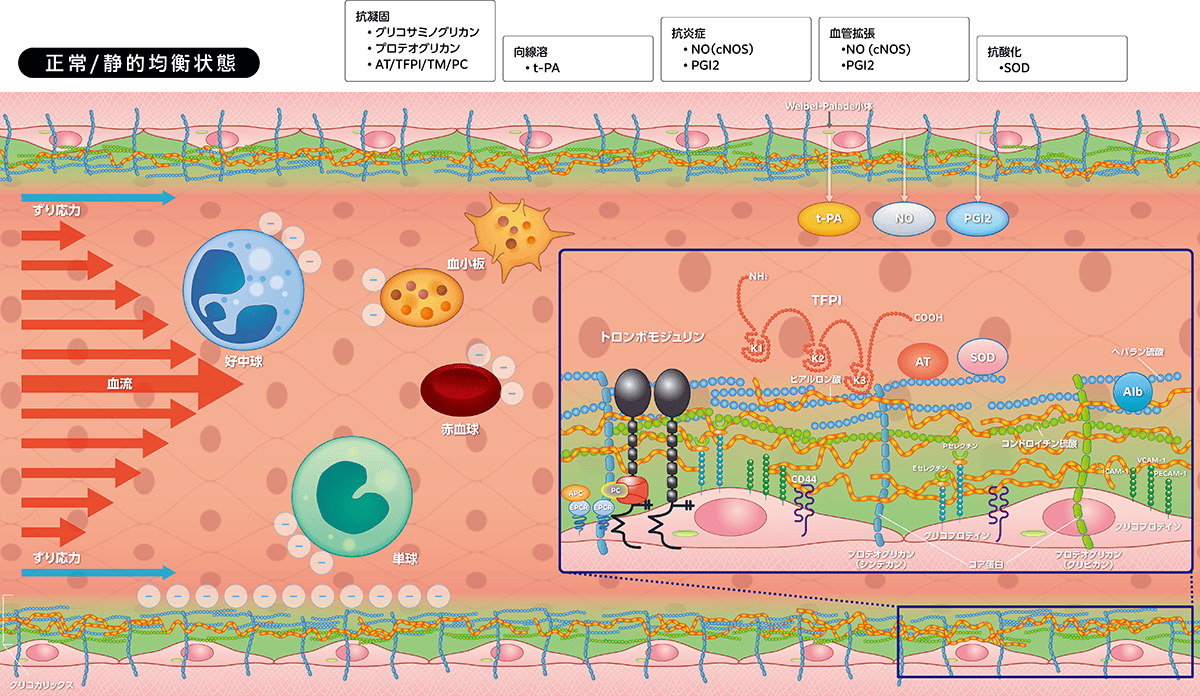
1)グリコカリックス発現細胞
グリコカリックスは毛細血管、小動静脈、中・大血管など、すべての血管に発現しますが、毛細血管で最も発達し血管内皮細胞と協働して重要な役割を担います。血管以外には、赤血球、白血球(単球、好中球、リンパ球)、血小板、そして気道・消化管・尿細管上皮細胞にも存在します。
2)グリコカリックス構造
グリコカリックスは、グリコプロテイン(セレクチン、インテグリン、免疫グロブリンスーパーファミリーなど)、プロテオグリカン(シンデカン、グリピカンなど)、グリコサミノグリカン(ヘパラン硫酸、コンドロイチン硫酸、ヒアルロン酸など)で構成されます。グリコプロテインは細胞接着分子群です。シンデカン(1〜4)、グリピカン(1〜6)にはサブタイプが存在し、おのおのの血管内皮細胞貫通コア蛋白にヘパラン硫酸とコンドロイチン硫酸が付着します。ヒアルロン酸はコア蛋白に付着せず、周りを漂いまとわりつくように存在していますが、その受容体CD44で血管内皮細胞に固定されます。
3)血管内皮細胞とグリコカリックスの生理作用
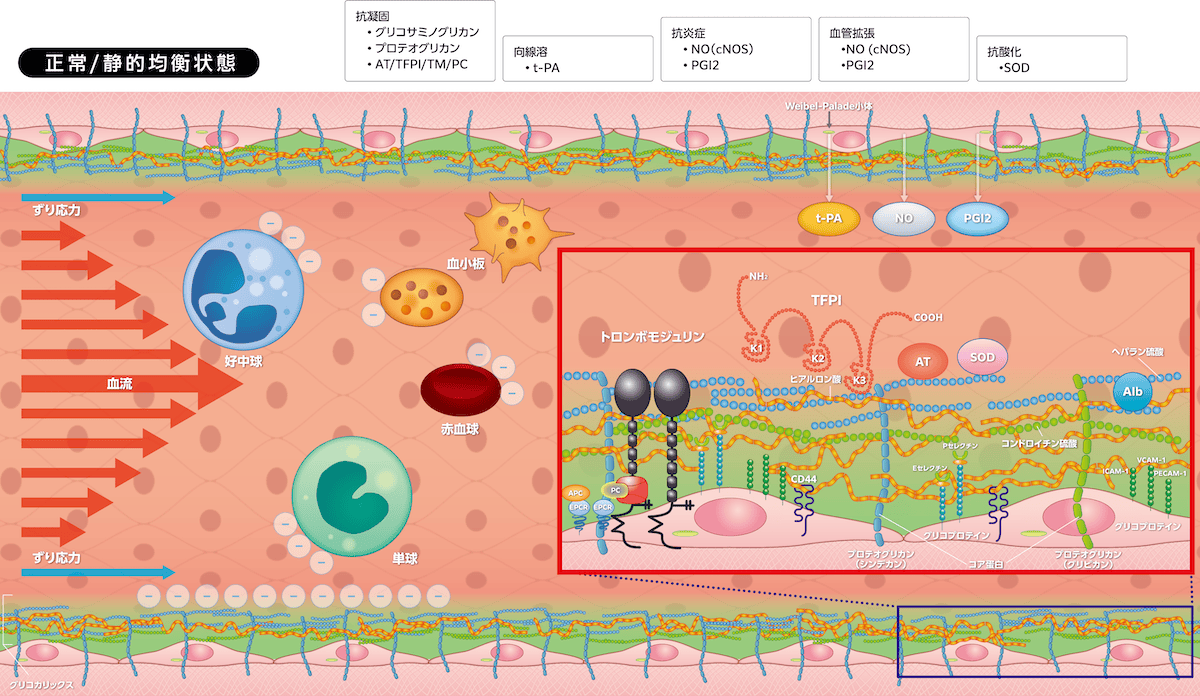
・抗凝固、向線溶活性
プロテオグリカン/ヘパラン硫酸には生理的抗凝固因子であるアンチトロンビンとTFPIが付着しています。アンチトロンビンは凝固因子(トロンビン、TF/FVIIa、FIXa、FXa、FXIa、FXIIa)とカリクレインを、TFPIは凝固因子(TF/FVIIa、FXa)を阻害します。血管内皮細胞に発現するトロンボモジュリンはトロンビンと複合体を形成して、活性化プロテインCを介してFVa、FVIIIaを分解・阻害して凝固反応を制御します。Weibel-Palade小体から放出されるt-PAはプラスミノゲンをプラスミンに変換して線溶活性が亢進します。このように血管内は抗凝固・向線溶状態となり、さらにPGI2による血小板凝集抑制作用が加わり抗血栓性を維持しています。
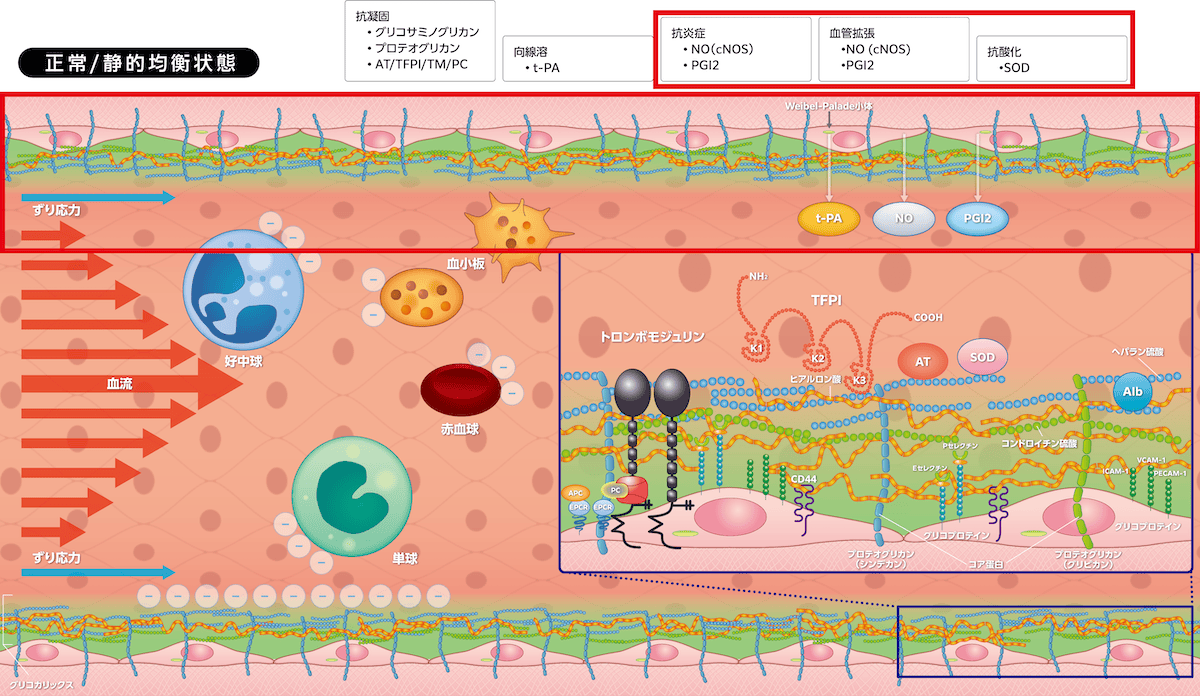
・抗炎症、抗酸化作用
ヘパラン硫酸はずり応力を関知し(mechanosensing)、プロテオグリカンを介して血管内皮細胞に情報を伝達します。その結果、血管内皮細胞に常時発現する
cNOS(eNOS)
cNOS:constitutive NO synthase
構成型一酸化窒素合成酵素。細胞内に常時発現している。存在場所で分類され、血管内皮細胞はeNOS、神経細胞はnNOSと呼称する。
がリン酸化して活性化、産生されたNOが血管拡張、血小板凝集抑制(抗血栓)、白血球接着抑制(抗炎症)を起こします。ずり応力は血管内皮細胞
PLA2
PLA2:phospholipase A2
ホスホリパーゼA2。アラキドン酸生成酵素で、プロスタグランジン(PG)、ロイコトリエン(LT)合成の起点となる。
を活性化してPGI2産生を誘導しますが、アンチトロンビンもシンデカン4を介してPGI2産生を誘導します(Vol.2-図1参照)。
 Vol.2 エキスパート版ページを開く
PGI2は血管拡張と血小板凝集抑制、白血球(好中球、単球)遊出抑制に寄与します。ずり応力で活性酸素種(ROS)が生じますが、ヘパラン硫酸に結合しているSODの抗酸化作用で過剰な産生が制御されます。
Vol.2 エキスパート版ページを開く
PGI2は血管拡張と血小板凝集抑制、白血球(好中球、単球)遊出抑制に寄与します。ずり応力で活性酸素種(ROS)が生じますが、ヘパラン硫酸に結合しているSODの抗酸化作用で過剰な産生が制御されます。
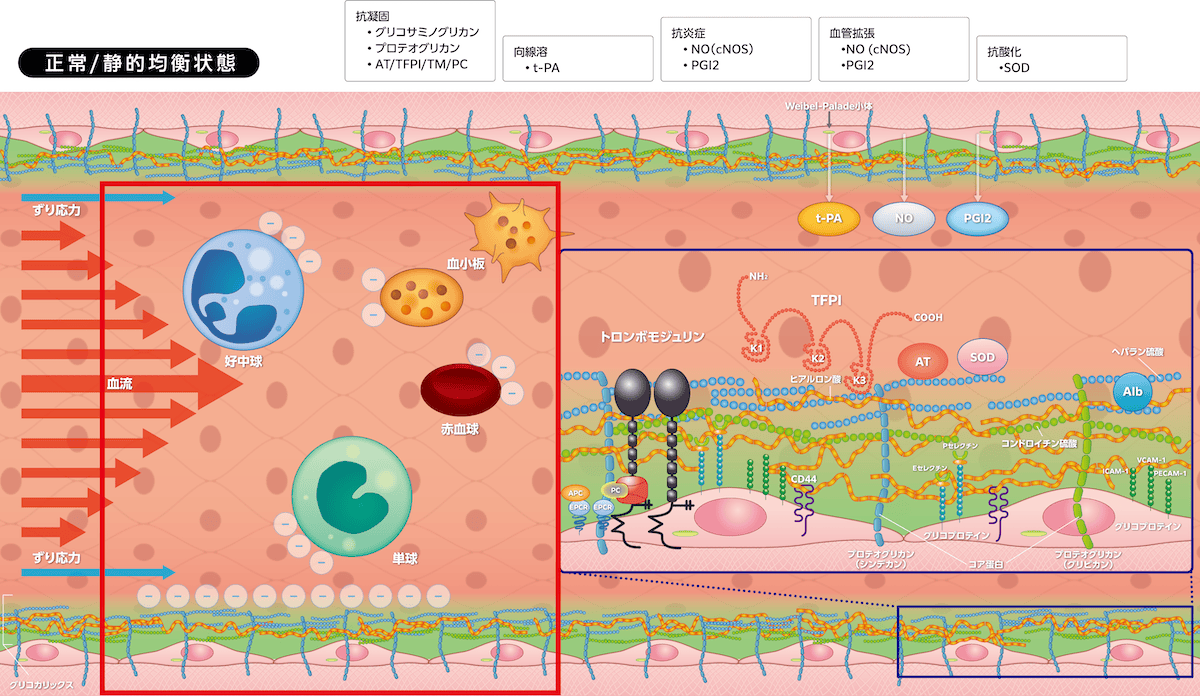
・陰性荷電と血液細胞
ヘパラン硫酸、コンドロイチン硫酸は陰性に荷電し、陰性荷電をもつ赤血球・白血球・血小板と相互作用し、赤血球の血管壁との接触回避・流動性保持、白血球の不要な接着防止・抗炎症作用、血小板粘着を抑制して抗血栓性を維持します。これらの血液細胞が活性化して露出するフォスファチジルセリンも陰性に荷電しており、グリコカリックスはフォスファチジルセリンの凝固活性を制御しています。
2.炎症反応とDIC
サマリー
侵襲によって起こる生理的炎症反応では、白血球・血管内皮細胞の活性化が起こり、血管内皮細胞上での白血球の回転・膠着・接着から血管外遊出に至りますが、この過程には血管内皮細胞接着分子の発現が関与します。一方、病原体関連分子パターン(
PAMPs
PAMPs:pathogen-associated molecular patterns
病原体関連分子パターン。非自己(リガンド)としてPRRsに作用する。
)/ダメージ関連分子パターン(
DAMPs
DAMPs:damage-associated molecular patterns
ダメージ関連分子パターン。変容した自己(リガンド)としてパターン認識受容体(pattern recognition receptor; PRRs)に作用する。
)やプロテアーゼがグリコカリックスを分解・脱落させ、また血管内皮細胞傷害を引き起こし、向凝固や抗線溶、向炎症・向酸化などの病的炎症反応が起こり、DICを発症します。
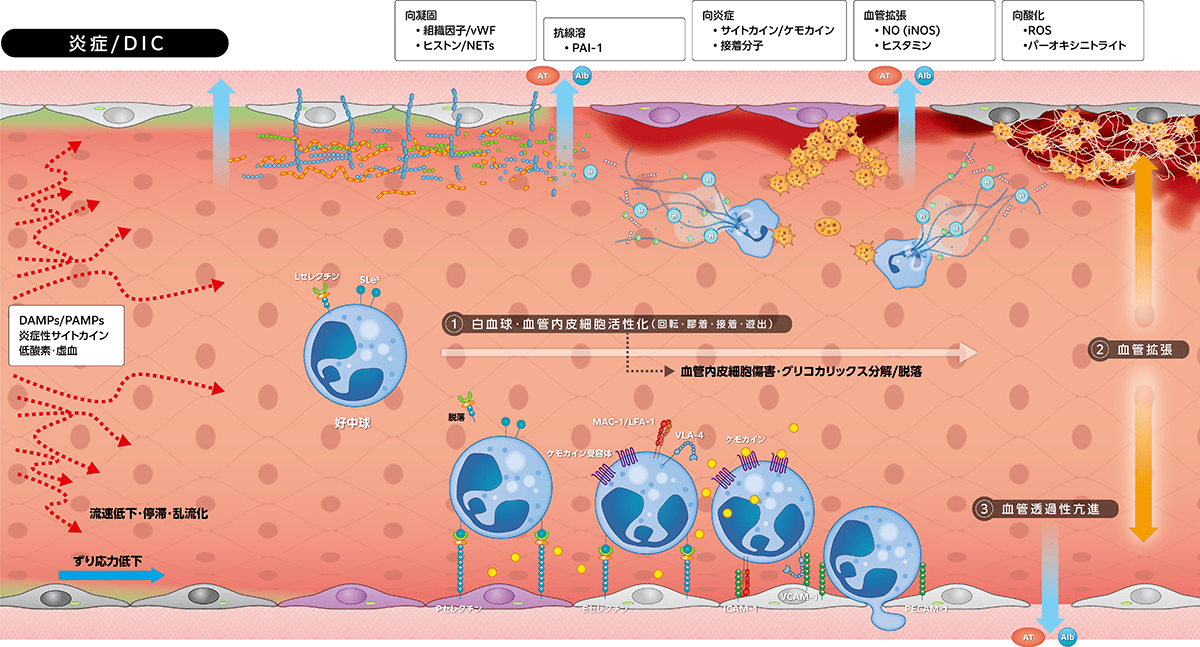
1)生理的炎症反応
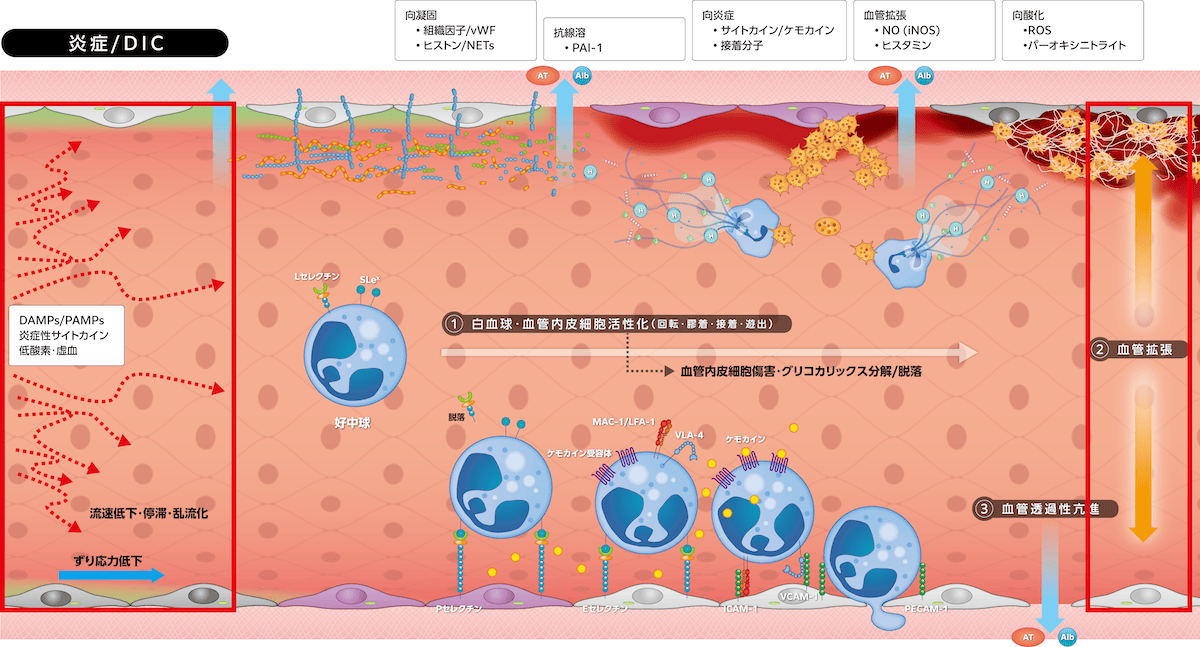
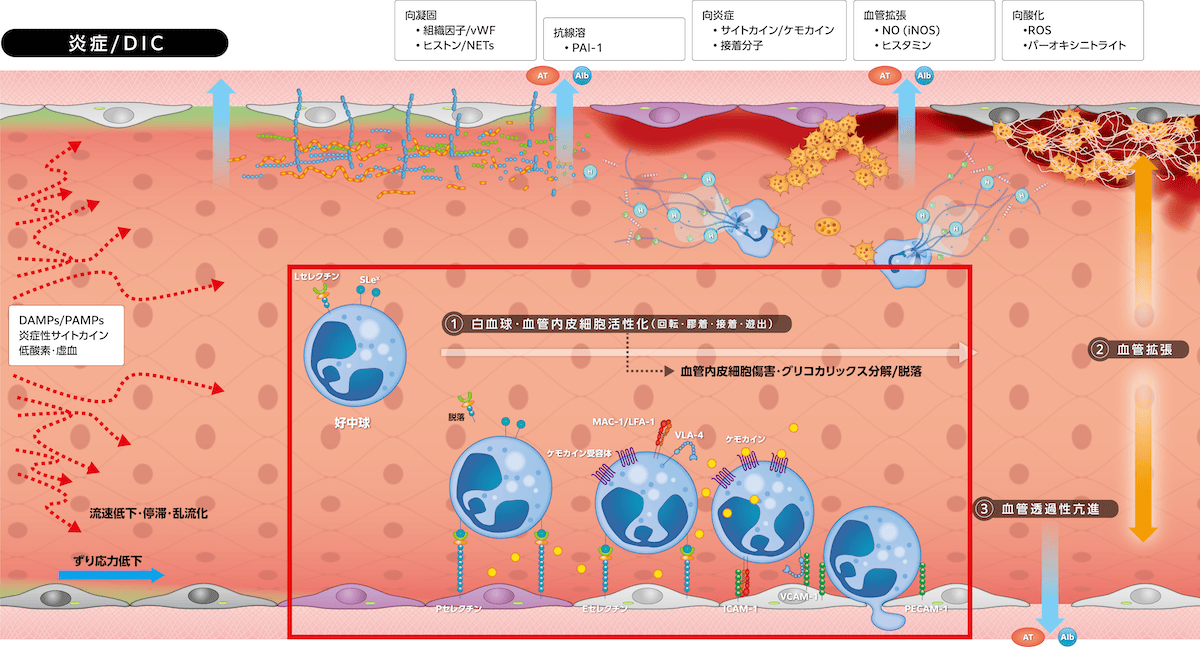
生体侵襲刺激(PAMPs/DAMPs、炎症サイトカインなど)は血管内皮細胞にiNOS発現を誘導してNO
産生が亢進し、PGI2とともに血管を拡張します。同時にヒスタミン、ブラジキニンなどが血管透過性を亢進させ、さらに発痛により侵襲の存在を生体に知らせます。
引き続き白血球(図は好中球)および血管内皮細胞が活性化し、白血球が血管内皮細胞上を回転(rolling)・膠着(tethering)・接着(adhesion)し、その後血管外遊出(transmigration)しますが、この過程に細胞接着分子群(グリコプロテイン)が関与します。血管内皮細胞Weibel-Palade小体に貯蔵されているPセレクチンは刺激後数分で血管内皮細胞上に露出し、その後Eセレクチンが発現し4~6時間後に最高値となり、十数時間で免疫グロブリンスーパーファミリー(
ICAM-1
ICAM-1:intercellular adhesion molecule-1
細胞接着分子-1。免疫グロブリンスーパーファミリーに属するグリコプロテイン。
、
VCAM-1
VCAM-1:vascular cell adhesion molecule-1
血管細胞接着分子-1。免疫グロブリンスーパーファミリーに属するグリコプロテイン。
)の発現が誘導されます。白血球上のLセレクチンは白血球活性化に伴い
MMP9
MMP:matrix metalloprotease
好中球マトリックスメタロプロテアーゼ。特殊顆粒内に存在する。
で分解され脱落(shedding)します。白血球は細胞表面糖鎖
SLeX
SLeX:Sialyl Lewis X
白血球表面糖鎖。
を介してPセレクチン・Eセレクチンと結合して回転を開始し、ケモカイン(IL-8、
MIP-1α
MIP-1α:macrophage inflammatory protein 1α
マクロファージ炎症性タンパク質。ケモカインの一種。
など)で活性化したインテグリン(
MAC-1
MAC-1:Macrophage-1 antigen
マクロファージ抗原-1。細胞接着分子インテグリンの一種。
、
LFA-1
LFA-1:lymphocyte function associated antigen
リンパ球機能関連抗原。細胞接着分子インテグリンの一種で、リンパ球やその他の白血球上に存在する。
、
VLA-4
VLA-4:very late antigen-4
最晩期抗原-4。細胞接着分子インテグリンの一種。
)を介してICAM-1、VCAM-1と結合・膠着し、血管内皮細胞へ接着していきます。接着した白血球は侵襲局所へ血管内皮細胞間隙から
PECAM-1
PECAM-1:platelet endothelial cell adhesion molecule-1
血小板内皮細胞接着分子。免疫グロブリンスーパーファミリーに属する細胞表面レセプター。
を介して浸潤・遊出して侵襲(病原微生物など)と遭遇します。
炎症の三主徴は「白血球・血管内皮細胞活性化、血管拡張、血管透過性亢進」であり、これらが侵襲局所では「発赤・腫脹・疼痛・熱感」として発現し、全身へ波及すると「頻脈・頻呼吸・発熱・白血球増多」としてSIRSを呈します。
2)病的炎症反応とDIC
PAMPs(
LPS
LPS:lipopolysaccharide
リポ多糖。グラム陰性菌外膜構成因子でpattern-associated molecular patterns(PAMPs)として作用する。
など)/DAMPs(
ヒストン
ヒストン:histones
DNAを巻き付けてヌクレオソームを形成する。ヌクレオソームが集合しクロマチンを形成し核内にDNAをコンパクトに収納している。
、
HMGB1
HMGB1:high-mobility group box-1
細胞核内に存在するdamage associated molecular pattern(DAMPs)で、終末糖化産物受容体(RAGE)を受容体とする。
など)、炎症性サイトカイン(TNFα、IL1β、IL6など)は白血球(好中球・単球)を活性化し細胞内顆粒からヘパリナーゼとMMP9を放出、
NETs
NETs:neutrophil extracellular traps
好中球細胞外トラップ。DNA、ヒストンで構成され、好中球エラスターゼなどが付着するクロマチン網。
にはヒストン、エラスターゼ・カテプシンGが付着、さらに血管内皮細胞にヘパリナーゼとMMP7を発現誘導します。PAMPs/DAMPsが直接、あるいは産生放出されたプロテアーゼがプロテオグリカン・グリコサミノグリカンを分解・脱落し、血管内皮細胞を傷害して病的炎症反応が起こります。
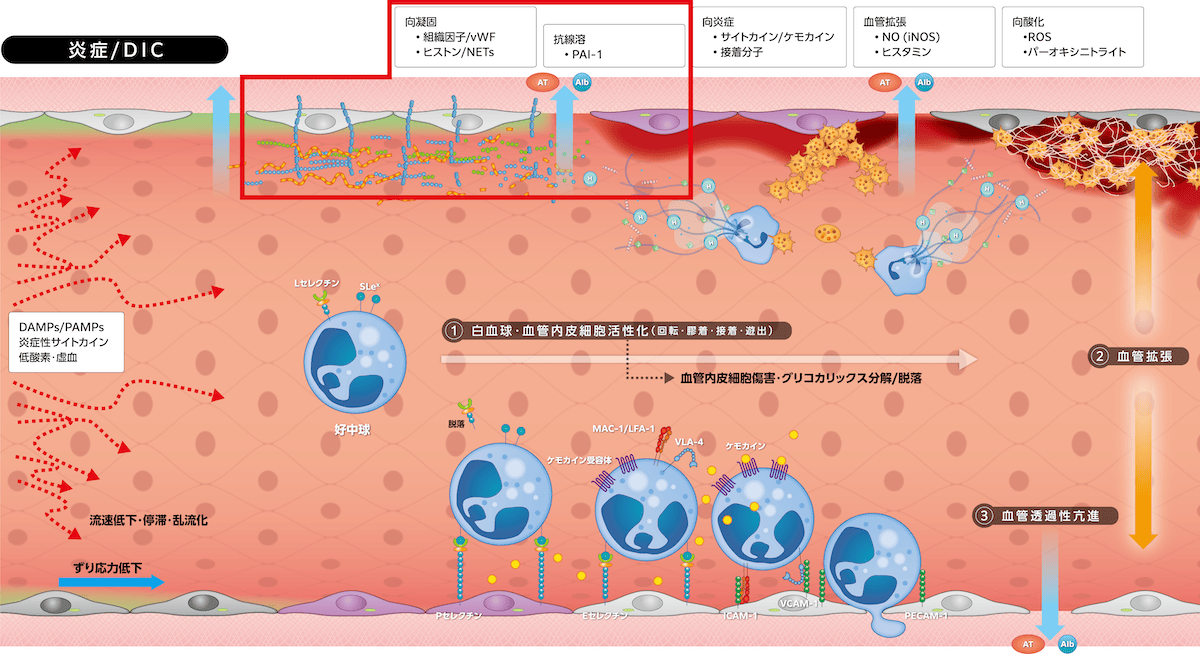
・向凝固、抗線溶反応
生体侵襲刺激は白血球・血管内皮細胞の組織因子発現と血管内皮細胞から
vWF
vWF:von Willebrand Factor
血管内皮細胞で産生、Weibel-Palade小体に貯蔵される。血管損傷に伴い同小体から放出され血小板一次止血に関与する。
放出を増強し、血小板活性化・凝固を亢進させ、同時に血管内皮細胞に抗線溶因子
PAI-1
PAI-1:plasminogen activator inhibitor-1
t-PAと結合して不活性化する線溶抑制因子。
を発現します。しかし、グリコカリックス分解・脱落でアンチトロンビンとTFPIの機能が減弱し、血管内皮細胞傷害に伴いトロンボモジュリンも分解・脱落し、プロテインC/活性化プロテインC変換が抑制されます。血管透過性亢進はアンチトロンビンの血管外漏出を促進し、凝固亢進に伴いアンチトロンビン・プロテインC・TFPIは消費性に減少します。これらの結果、凝固亢進とフィブリン血栓形成に見合う抗凝固・向線溶機能(t-PA)が働かずDICが発症します。
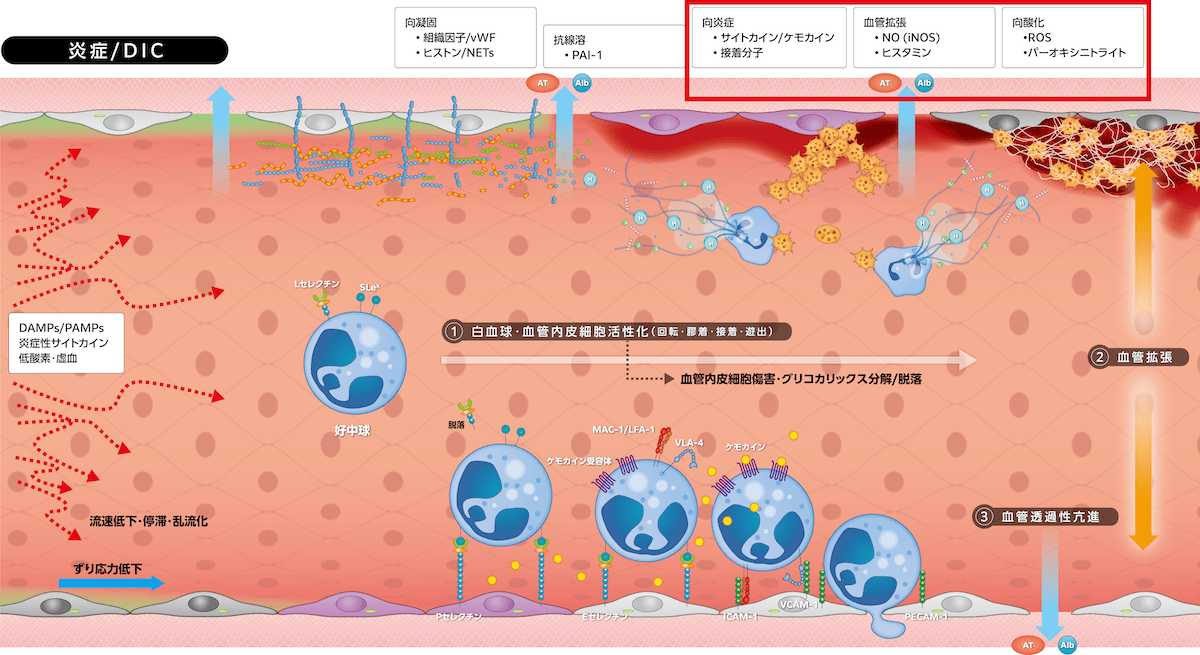
・向炎症、向酸化作用
白血球活性化に伴う炎症性サイトカイン発現増強、プロテアーゼ放出、活性酸素種産生が炎症を増強します。グリコカリックス分解・脱落による陰性荷電バリアの消失と、その菲薄化によるグリコカリックス外への接着分子の露出に伴い、白血球接着が加速・促進されて炎症反応がさらに増強します。ヘパラン硫酸に結合しているSODが血中に逸脱し炎症局所の抗酸化作用が低下するために、産生された活性酸素種分解が減少、スーパーオキシド(O2-)がNOと反応しパーオキシニトライト(ONOO-)が形成されて酸化的ストレス傷害が起こります。
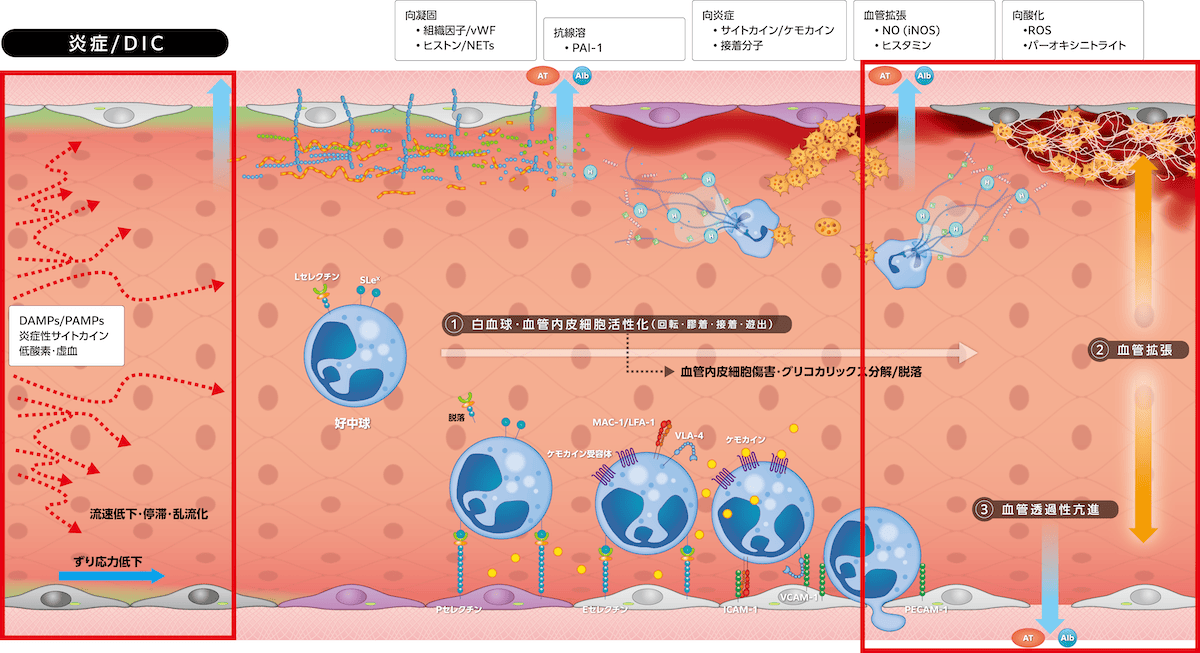
・血管拡張と透過性亢進
炎症反応初期には誘導されたiNOSがNO産生を亢進して血管拡張が起こります。しかし、グリコカリックス分解・脱落によるずり応力関知機能低下がeNOS活性低下かつiNOS発現を減少して血管拡張機能が低下します。同時にカドヘリンなどの細胞間隙接着分子機能が低下して血管透過性が亢進し、ヘパラン硫酸を介してグリコカリックス内に保持されていたアルブミン、アンチトロンビンが漏出します。グリコカリックス分解・脱落は血流へも影響し、ずり応力低下、乱流化、逆行・偏流化、流速低下・停滞が起こり、ずり応力依存性の生理機能が障害される悪循環が形成されます。
3.炎症・低酸素連関(inflammatory hypoxia)とDIC
サマリー
DICでは炎症による酸素消費増大と酸素供給減少で発現する低酸素誘導因子(HIF1α)が重要な役割を担い、
アンジオポエチン(Ang)
アンジオポエチン:angiopoietin(Ang)
血管新生・血管内皮細胞間接着関連因子。
/Tie2が主役となります。正常状態ではAng/Tie2が細胞間隙細胞接着の増強、組織因子発現・
フォスファチジルセリン
フォスファチジルセリン:phosphatidylserine
細胞内Ca2+上昇で活性化したスクランブラーゼで、血小板等のリン脂質膜表面に露出し凝固因子を活性化する。
露出・接着分子発現の制御に働くのに対し、炎症・低酸素下の病的状態ではHIF1αの発現増強からAng2・
VEGF
VEGF:vascular endothelial growth factor
血管内皮細胞増殖因子。血管新生因子の一種。
、炎症性サイトカイン、組織因子・PAI-1の発現が誘導され、TFPI・
プロテインS
プロテインS:protein S(PS)
APCはPSを補酵素としてFVa、FVIIIaを分解不活化する。
の発現が減少するほか、Ang/Tie2が細胞間隙細胞接着を脆弱化させ、血管透過性が亢進し、組織因子発現・フォスファチジルセリン露出・接着分子発現・NETs放出を促進します。
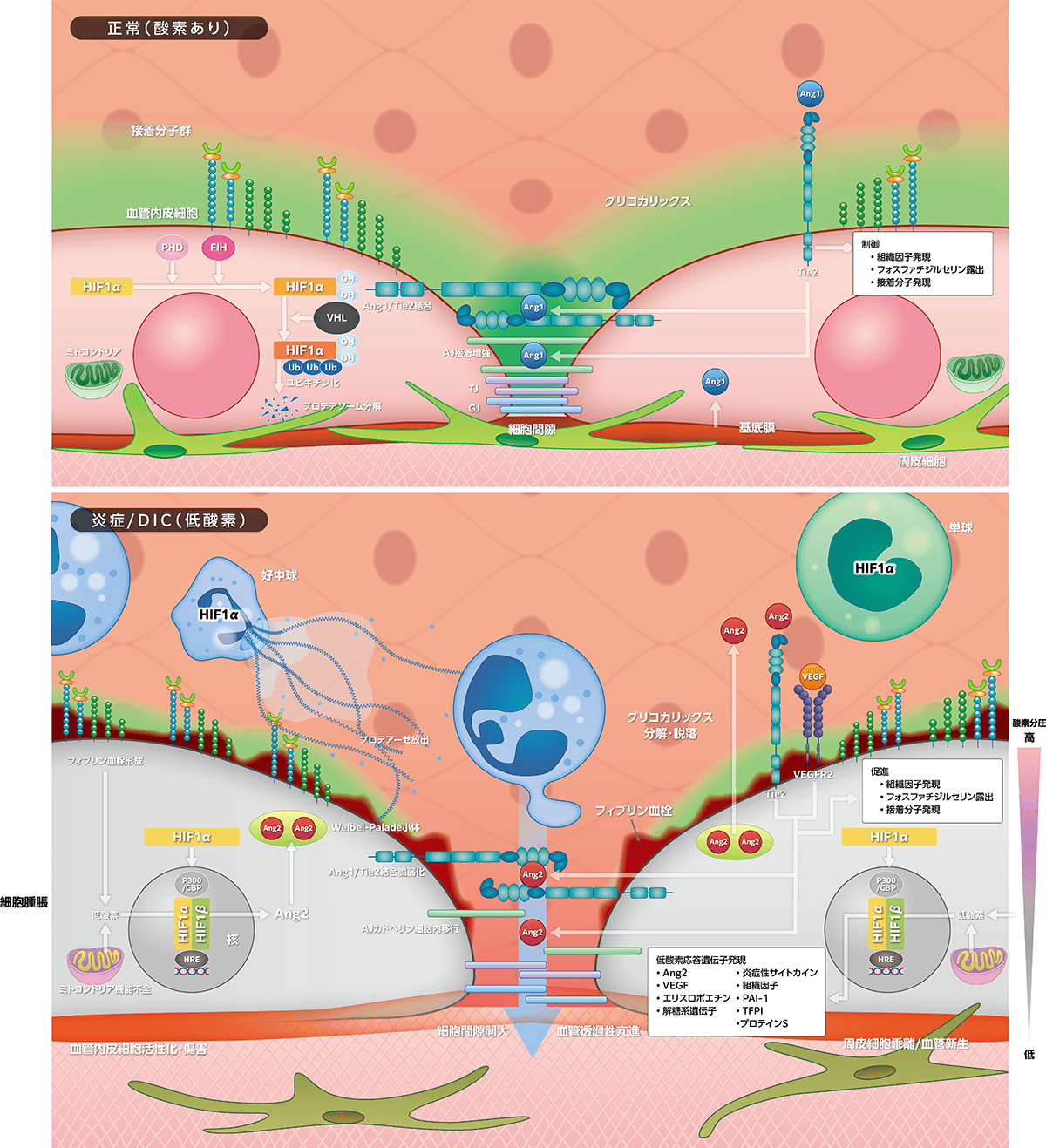
炎症細胞では酸素消費が増加しますが、炎症局所に集簇する活性化白血球(好中球・単球)も酸素消費が増しています。さらに、炎症に伴う凝固亢進による血栓形成が酸素運搬を障害します。酸素供給減少と酸素消費増加が酸素負債を生じ、血管・細胞内の酸素分圧較差を助長して炎症細胞は低酸素に陥ります。恒常性維持のために低酸素誘導因子(HIF1α)により低酸素誘導遺伝子が発現し血管新生が起こりますが、その主役がAngiopoietin(Ang)/Tie2です。病的状態ではこれらが炎症・低酸素連関/DICのフィブリン血栓形成と血管透過性亢進に重要な役割を担います。
1)正常状態
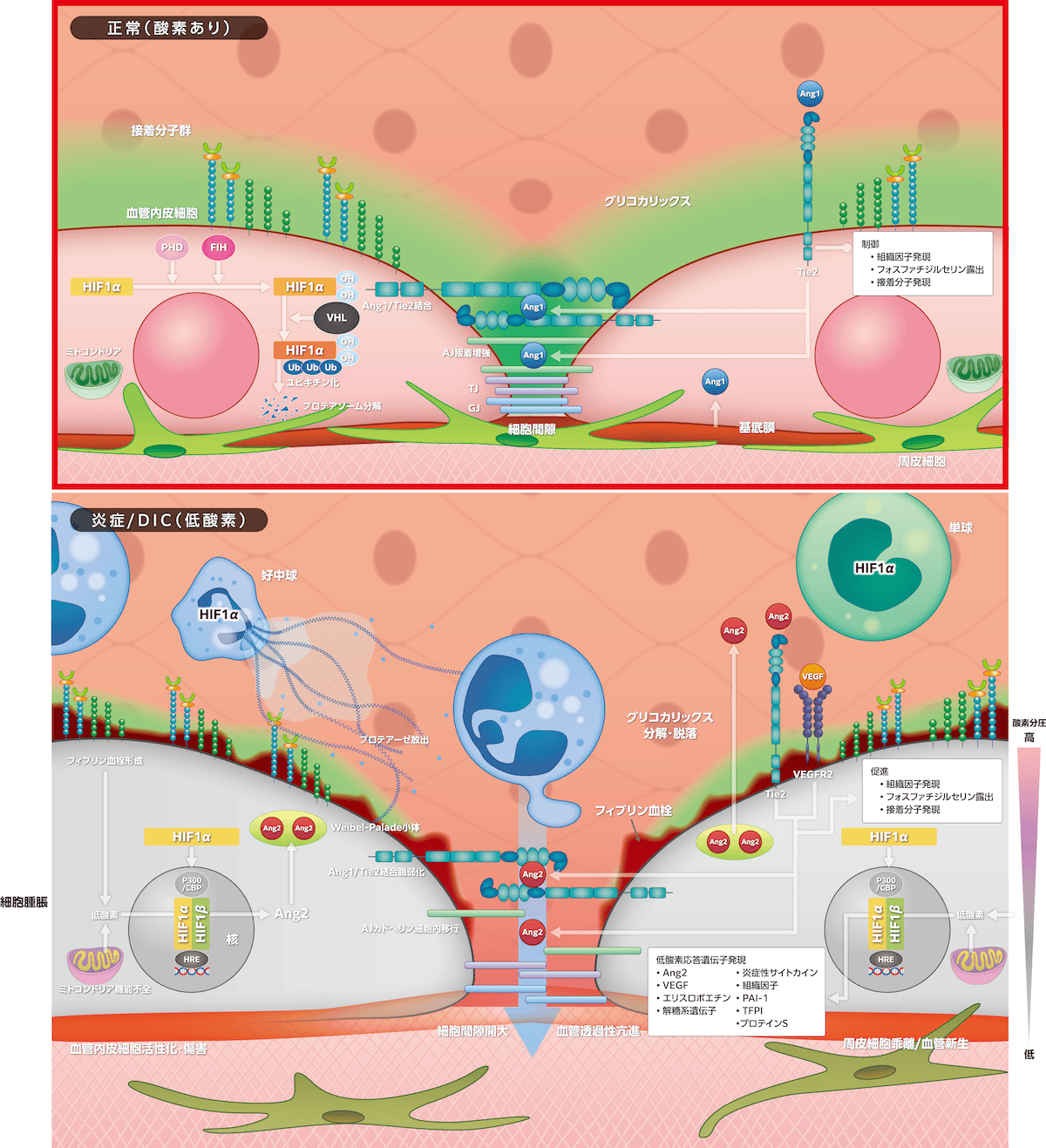
内皮細胞、基底膜、壁細胞(毛細血管では周皮細胞、その他では血管平滑筋細胞)、外膜が血管壁を構成しますが、壁細胞は内皮細胞を補強し安定化させる機能をもっています。血管内皮細胞間に細胞間隙が存在し、3つの細胞間接合(
AJ
AJ:adherences junction
血管内皮細胞間隙接合を司る。
、
GJ
GJ:gap junction
血管内皮細胞間隙接合を司る。
、
TJ
TJ:tight junction
血管内皮細胞間隙接合を司る。
)とグリコカリックスが血管透過性を制御しています。
正常酸素化では、ミトコンドリア呼吸の余剰酸素が
PHD
PHD: prolyl hydroxylase
HIF-1αを水酸化して、その分解に関与する。
と
FIH
FIH: factor inhibiting HIF-1α
HIF抑制因子。酸素依存性に機能し、正常酸素化ではHIF-1αのP300/CBPへの結合を阻害する。
に作用しこれらが正常に機能します。HIF-1αはPHD作用で水酸化し、
VHL
VHL: von Hippel-Lindau protein
ユビキチンリガーゼと会合しHIF-1αをユビキチン化する。
ユビキチンリガーゼ作用を受けて
ユビキチン化
ユビキチン化:Ubiquitination
特定のタンパク質にユビキチンが結合する反応。ユビキチン化されたタンパク質はプロテアソームによって認識され、分解を受ける。
(ユビキチンが結合)してプロテアソームで分解されます。さらにFIHがHIF-1αの核内P300/
CBP
CBP: cytoplasmic polyadenylation element binding (CREB)- binding protein
転写因子CREBと共役して機能する遺伝子発現活性化因子。
への結合を阻害するために、HIF-1αによる低酸素応答遺伝子発現が制御されます。
Ang1は壁細胞(周皮細胞、平滑筋細胞)で産生・分泌され、Ang2は血管内皮細胞で産生されWeibel-Palade小体に貯蔵された後に放出されます。Ang1とAng2の受容体が血管内皮細胞に発現するTie2であり、おのおのTie2へ刺激・抑制情報を伝達します。Ang1はTie2に結合し、血管内皮細胞間隙で細胞間を架橋して接合し、さらにAng1はグリコカリックスとともにAJ構成蛋白のカドヘリン接着を促進して血管透過性亢進を制御しています。Ang1/Tie2は血管内皮細胞の組織因子発現、フォスファチジルセリンの血管内露出を制御して凝固を抑制しますが、接着分子発現制御による抗炎症作用も併せもち、血管の静的均衡維持に貢献しています。
2)炎症・低酸素連関とDIC
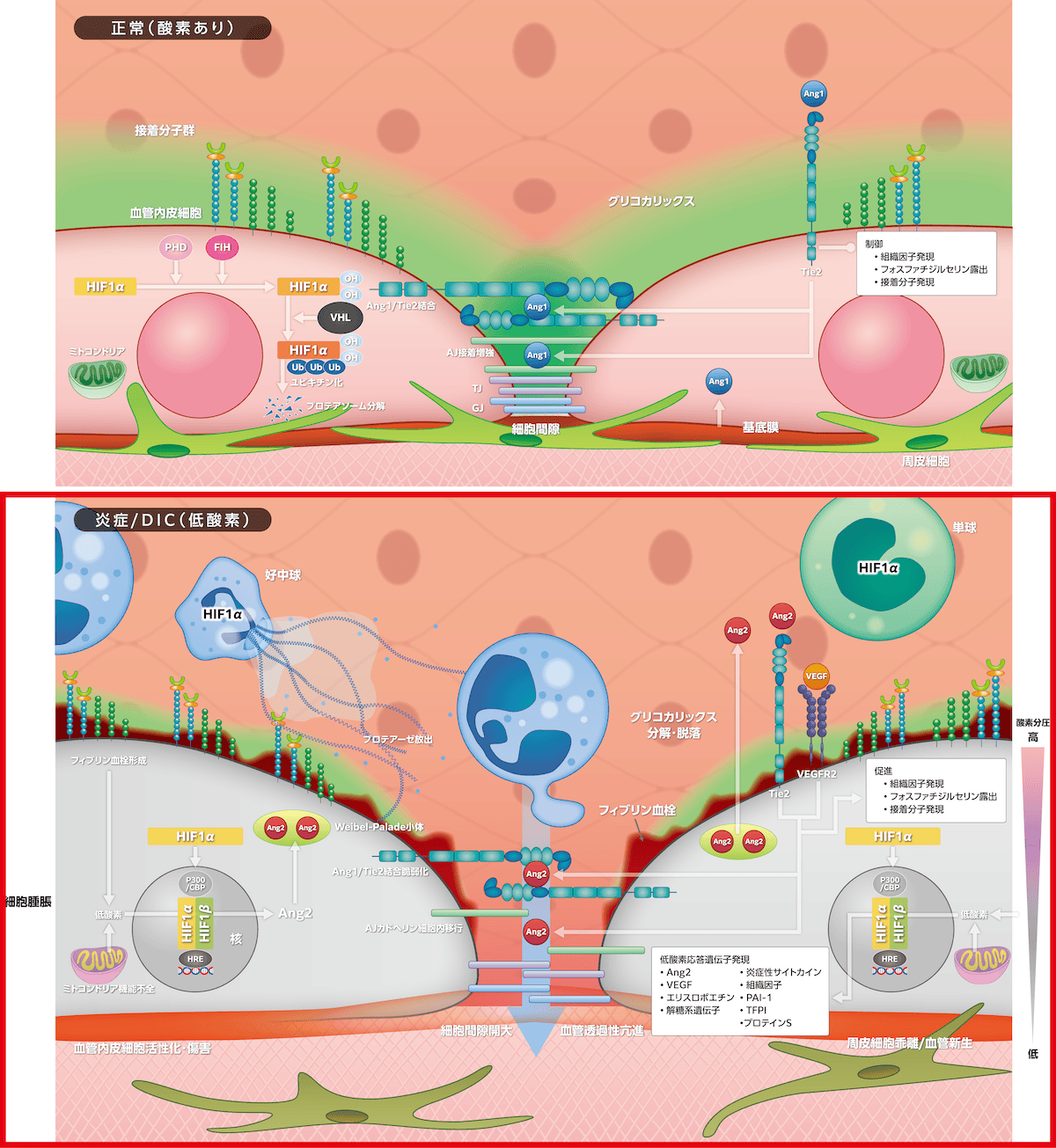
DICではフィブリン血栓形成下の低酸素、組織酸素代謝失調によるミトコンドリア機能障害による低酸素、そして炎症に伴う細胞内酸素分圧低下によりPHDが機能せずHIF1αのプロテアソーム分解が減少して、その発現が増強します。核内移行したHIF1αはFIH作用低下に伴いP300/CBPを介してHIF1βと会合して低酸素応答遺伝子Ang2とVEGF発現を誘導します。両者は周皮細胞乖離を通じて生理的血管新生作用をもつのですが、DICでは過剰発現して血管透過性亢進、凝固・炎症反応亢進へ関与します。Ang2はTie2に結合して、細胞間隙のAng1/Tie2結合を脆弱化し、AJ構成蛋白のカドヘリン蛋白を細胞内移行させて細胞間隙開大が起こります。同様に、VEGFは
VEGFR
VEGFR:VEGF receptor
VEGF受容体。
2に結合してカドヘリンの細胞内移行を促進して細胞間隙を拡大します。さらに、グリコカリックスの分解・脱落による陰性荷電消失は活性化白血球の細胞間隙侵入を容易とし、プロテアーゼがカドヘリンを不安定化します。細胞間隙拡大は血管透過性亢進をきたし、アルブミン・水分に加えてアンチトロンビン、フィブリノゲンなどの漏出が起こります。
Ang2はAng1に拮抗して組織因子発現、フォスファチジルセリン露出、接着分子発現を増強して凝固炎症反応を促進します。HIF1αは白血球にも発現して炎症性サイトカイン産生、組織因子発現、NETs形成を促進していますが、血管内皮細胞ではPAI-1発現を誘導して線溶を抑制し、凝固制御因子TFPIやプロテインS発現を抑制する作用もあります。このように、炎症・低酸素連関はDICの発症に加えて悪化要因として重要な役割を担います。
(審J2605037)

